Publications & Posters
Systemic Type I Ifn Inflammation In Human Isg15 Deficiency Leads To Necrotizing Skin Lesions
CELL REPORTS
Martin-Fernandez M, Bravo García-Morato M, Gruber C, Murias Loza S, Malik MNH, Alsohime F, Alakeel A, Valdez R, Buta S, Buda G, Marti MA, Larralde M, Boisson B, Feito Rodriguez M, Qiu X, Chrabieh M, Al Ayed M, Al Muhsen S, Desai JV, Ferre EMN, Rosenzweig SD, Amador-Borrero B, Bravo-Gallego LY, Olmer R, Merkert S, Bret M, Sood AK, Al-Rabiaah A, Temsah MH, Halwani R, Hernandez M, Pessler F, Casanova JL, Bustamante J, Lionakis MS and Bogunovic D
Cell Rep. 2020 May 12;31(6):107633
DOI: https://doi.org/10.1016/j.celrep.2020.107633
Highlights
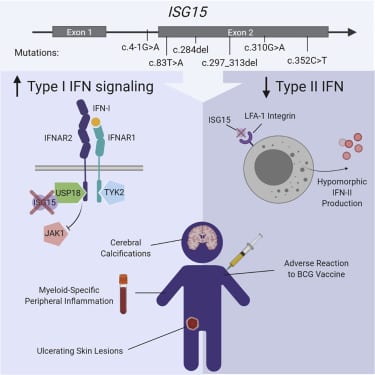
- ISG15 deficiency is identified in five new patients with six novel genetic lesions
- All patients presented with dermatologic complications
- ISG15 mutations lead to hyper-activated responses to type I interferon
- Patient cells exhibit striking cell type specificity to the interferon response
Summary
Most monogenic disorders have a primary clinical presentation. Inherited ISG15 deficiency, however, has manifested with two distinct presentations to date: susceptibility to mycobacterial disease and intracranial calcifications from hypomorphic interferon-II (IFN-II) production and excessive IFN-I response, respectively. Accordingly, these patients were managed for their infectious and neurologic complications. Herein, we describe five new patients with six novel ISG15 mutations presenting with skin lesions who were managed for dermatologic disease. Cellularly, we denote striking specificity to the IFN-I response, which was previously assumed to be universal. In peripheral blood, myeloid cells display the most robust IFN-I signatures. In the affected skin, IFN-I signaling is observed in the keratinocytes of the epidermis, endothelia, and the monocytes and macrophages of the dermis. These findings define the specific cells causing circulating and dermatologic inflammation and expand the clinical spectrum of ISG15 deficiency to dermatologic presentations as a third phenotype co-dominant to the infectious and neurologic manifestations.
Graphical Abstract